
Identification of cells by flow cytometry is typically accomplished using fluorescently labeled antibodies to specific targets on the surface of the cell. Researchers typically name the cells based on these targets, often referred to as Clusters of Differentiation or CD’s. For example, one of the components of the T-cell receptor is CD3, so researchers will often use antibodies to this target to identify this lineage and separate it from other cells that do not express the protein.
Stepping back to the basics of immunology, an antibody can be visualized as a ‘Y’ shaped protein. The backbone is made up of two heavy chains that are linked by disulfide bonds. On either side of the Y fork are two additional proteins called ‘light chains’, which are also bound by disulfide bonds to this backbone. The active site of the antibody is at the tip of the Y, the specificity being determined by both the heavy and light chain.
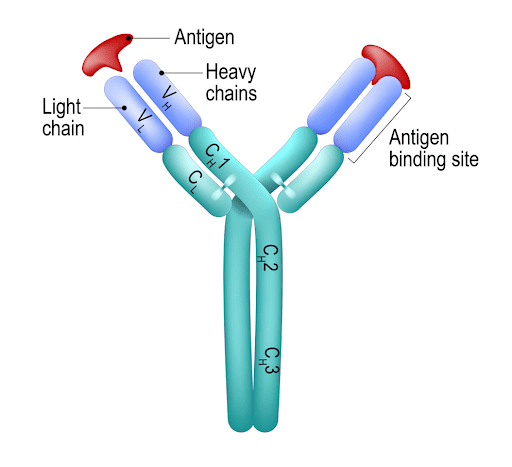
Figure 1: The structure of an antibody. The binding site is noted on the left arm
The heavy and light chains arise from the combining of different segments in the DNA. In the case of the variable region of the heavy chain, this includes three units – the Variable, the Diversity, and the Joining segments. The light chain only has two units, the Variable and Joining segments. These are then joined to the constant segments to make the complete peptide. This is what leads to the vast array of antibodies in the body.
However, this is just the beginning and the processes to refine these antibodies involve somatic hypermutation, which induces point mutations in the variable domains that can impact the antibodies’ specificity and affinity.
To make matters more complex, these antibodies can undergo a process called class switching. When an antibody is first made, the immature B-cell expresses the IgM form of the antibody. As the cell matures to become a naive B-cell, it expresses both the IgM and IgD form of the test antibody. These serve as the early warning system, ready to respond to new antigens.
We term these different forms ‘isotypes’, and each class of isotype has different biological properties in the body. The human heavy chain has five different heavy chain classes, while the light chain has two.
Antibodies to be used in research can either be polyclonal or monoclonal.
In flow cytometry, monoclonal antibodies are preferred due to monospecificity, consistency of production, and the like. However, there is always the concern that there can be non-specific binding by these antibodies on the target cells.
Enter the Isotype control. The idea behind the isotype control is that it is an antibody that has the same isotype as the experimental antibody, but has been produced against something that is not present on the cells of interest. For example, clone UCHT1, (mouse anti-human CD3) has an isotype of IgG1,κ. If there were concerns over non-specific binding of this antibody on the cells of interest, one might label a different tube with an isotype control such as MOPC-21, which is also isotype of IgG1,κ. This antibody was made from mouse myeloma, and has an unknown specificity. So the theory goes that if the MOPC-21 binds to the cells, the researcher would set the positive gate above staining levels of the isotope control, as shown in figure 2 below.
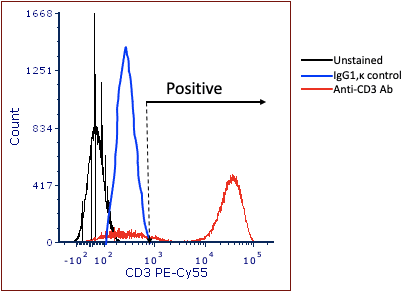
Figure 2: Using the isotype control to set ‘positivity’.
There are some assumptions and caveats that need to be considered when using the isotype control to set positivity, and the usefulness of the isotype control is heavily dependent on how much it violates these assumptions.
Taken together, violation of any of these three assumptions changes the isotype control into another experimental variable, thus eliminating its value. In fact, as early as 1999, clinical researchers were questioning the use of isotype control in analysis.1
More recently, an article by Andersen and coworkers reviewed this issue as well as ways to block Fc mediated binding on their target cells.2 They concluded that proper blocking was more important than using isotype controls, which was elegantly demonstrated due to variability in quality of isotope control that the researchers obtained commercially.
In conclusion, it should be noted that there is not a consensus for the use of the isotype control in flow cytometry. It is important to not overinterpret the isotype control, that is do not use it to set positivity. However, it is useful in showing that the blocking of cells was poor or incomplete, and when used in that manner, does provide some information to the researcher. However, most flow cytometrists have moved away from using these reagents in their experiments because of the limitations mentioned above.
Sources:





