Gentle Stem Cell Sorting Advances ALS Research

Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a fatal neurodegenerative disease marked by the progressive loss of motor neurons. As these neurons that control muscle movement die, patients lose the ability to walk, speak, swallow, and eventually breathe. While some ALS cases are inherited, most are sporadic, and the biological mechanisms driving disease remain only partially understood.
One of the most powerful tools to study ALS is the use of induced pluripotent stem cells (iPSCs). By reprogramming adult patient cells back into a stem cell state, researchers can generate motor neurons, astrocytes, and microglia that carry the same genetic mutations as the patient. These iPSC-derived models allow scientists to observe disease processes in human cells, which is something that was not possible with earlier animal models or immortalized cell lines. iPSCs also enable comparisons across different genetic subtypes of ALS, helping reveal both common and divergent disease mechanisms.
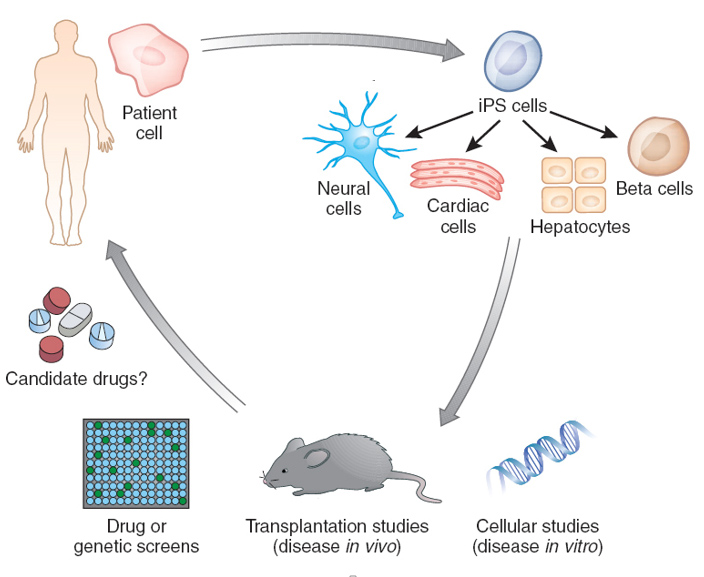
Figure 1. Pluripotent stem cells enable modeling of human disease in a petri dish. (Credit: Lorenz Studer Lab, Memorial Sloan Kettering)
Why Gentle Sorting Matters in ALS Research
The same qualities that make iPSCs so valuable also make them fragile. iPSC-derived neurons and glia are highly sensitive to stress, and even small perturbations from conventional high-pressure cell sorting can alter their gene expression, viability, or function. This means that without gentle, precise isolation, researchers risk losing the very disease signatures they are trying to measure. By preserving the health and authenticity of iPSC-derived cells, gentle cell sorting ensures that downstream discoveries truly reflect patient biology.
The WOLF and WOLF G2 solve this challenge by sorting cells at low pressure (<2 psi) through disposable microfluidic cartridges. This gentle approach ensures that fragile iPSC-derived neurons and glia remain healthy, enabling researchers to:
• Capture subtle disease signatures without stress artifacts
• Cleanly separate mixed populations for functional assays
• Maintain sterility for downstream experiments and drug testing
Two Studies, Two Mutations, Shared Insights
Recent research illustrates the power of this approach. In a 2024 study in Human Molecular Genetics, scientists investigated two of the most common genetic causes of ALS: mutations in SOD1, an enzyme that helps clear harmful free radicals, and C9ORF72, a gene involved in RNA processing and vesicle trafficking. In another study, researchers from Case Western Reserve University focused on a rarer but informative mutation in VAPB, a gene critical for maintaining communication between mitochondria and the endoplasmic reticulum.
At first glance, these genes have very different functions. Yet, when patient-derived iPSCs were differentiated into neurons and glia, common themes emerged: dysfunctional interactions between glial cells and neurons, disrupted mitochondrial activity, and heightened cellular stress responses. These findings underscore how iPSCs enable researchers to link diverse genetic mutations to shared biological pathways that may be targeted therapeutically — not only in ALS but in other neurodegenerative diseases as well.
Case Study 1 – Distinct Glial–Neuron Interactions in ALS
The Human Molecular Genetics study asked how astrocytes and microglia (two types of glial cells) contribute to motor neuron degeneration. Traditionally seen as “support cells,” glia are now recognized as active players in neurodegeneration, capable of either protecting or harming neurons depending on their state.
By generating astrocytes, microglia, and motor neurons from ALS patients with SOD1 or C9ORF72 mutations, researchers could study how these cell types interact. They found:
• C9ORF72 astrocytes produced high levels of the inflammatory molecule CCL2, which promoted harmful activation of surrounding cells and damaged motor neurons.
• SOD1 astrocytes did not show this effect on their own but became inflammatory when influenced by SOD1-mutant microglia.
When scientists boosted the anti-inflammatory signal IL-10 or blocked CCL2, they observed reduced glial activation and healthier motor neurons. These results suggest that, although the two mutations operate differently, they converge on inflammatory pathways that may be druggable targets. These important findings, a promising path for future ALS therapeutics, would have been masked if the iPSCs and the functioning of their differentiated glial cells had been altered in the process of cell isolation.
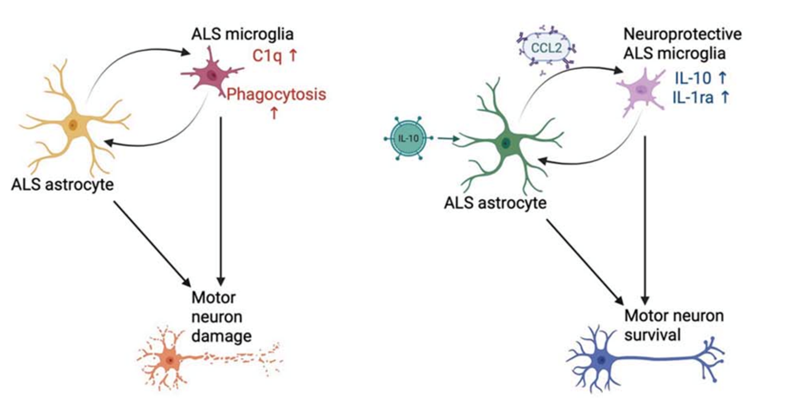
Figure 2. Schematic of ALS glial-mediated activation and motor neuron damage (left) and hypothesized effects of astrocyte-targeted treatments. Using iPSC-derived motor neurons, astrocytes, and microglia carrying ALS mutations, researchers showed that gentle sorting of these fragile cell types enabled clear observation of how astrocyte- and microglia-driven inflammation contributes to ALS pathology. (Credit: Allison & Ebert. 2024)
Case Study 2 – Mitochondrial Dysfunction in VAPB-Linked ALS
The second study examined ALS caused by a mutation in the gene that codes for VAPB, a protein that helps tether mitochondria to the endoplasmic reticulum (ER). This contact site is essential for energy production, calcium signaling, and cellular stress regulation.
Motor neurons derived from iPSCs expressing the VAPB P56S mutation exhibited:
• Reduced ER–mitochondria contact, leading to impaired communication between these organelles
• Declining mitochondrial membrane potential, a measure of energy-generating capacity
• Decreased firing and connectivity as the neurons matured, reflecting functional deficits
Crucially, the mutant neurons showed activation of the integrated stress response (ISR), a pathway that cells use to adapt to stress but which, when chronically engaged, can be toxic. Treatment with ISRIB, a small-molecule inhibitor of the ISR, rescued mitochondrial function and neuronal activity. This highlights mitochondrial dysfunction and stress signaling as central features of ALS pathogenesis.

Figure 3. Model of proposed pathogenesis in VABP mutant cells. Isolation of iPSCs by the WOLF Cell Sorter and a precise differentiation protocol of these biologically accurate cell lines elucidated, not just dysfunctional phenotypes, but also pinpointed when in the differentiation process the different responses seem to occur. (Credit: Landry et al. 2024)
From Basic Biology to Therapeutic Targets
Together, these ALS studies illustrate how iPSC-based models are revealing the cellular underpinnings of neurodegeneration. Whether it is astrocyte-driven inflammation in SOD1 and C9ORF72 ALS, or mitochondrial stress in VAPB-linked ALS, these insights point toward new therapeutic strategies.
With gentle, precise sorting from the WOLF platform, researchers can trust that their stem cell models reflect patient biology—not sorting-induced artifacts. The result is more reliable science, deeper biological insights, and faster progress toward treatments for ALS.


