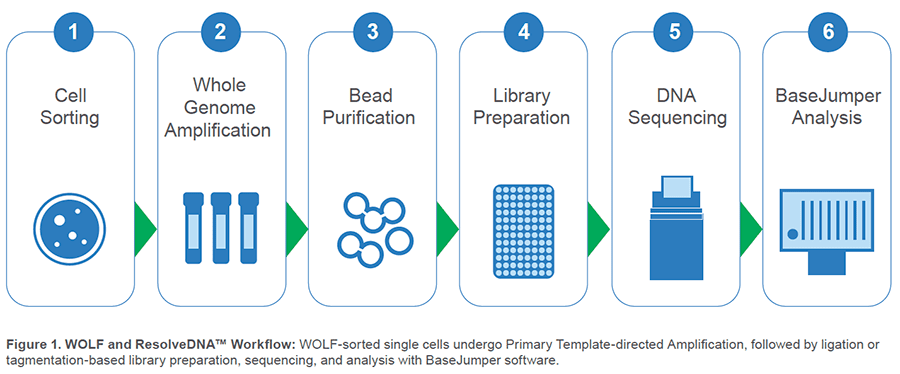
Whole-genome sequencing of single cells involves a multitude of technical challenges:
However, NanoCellect and BioSkryb have established a workflow to simplify this process and bypass these hurdles. This workflow profile highlights how next-generation cell sorting
and sequencing technologies can be used to obtain high quality single-cell genomic data.
Upstream, NanoCellect’s WOLF Cell Sorter:
Downstream, BioSkryb’s ResolveDNA™ platform:
EASILY fit the entire instrument into the smallest biosafety cabinets with <2 ft3 footprint. Master sorting quickly with user-friendly software, abundant training, and exceptional customer service.

CONFIDENTLY avoid contamination from previous experiments with disposable sterile cartridges and fluidics. When using pre-loaded well-specific reagents, switch from dispensing cells directly to the well bottom (Direct-Dispense Mode) to touching them off on the sidewall of each well (Sidewall-Dispense Mode) to avoid well-to-well crossover.
GENTLY sort cells-of-interest without shear force and with less than 2 PSI pressure. Avoid worrying about changes to cell viability and sequence quality.
ACCURATELY dispense up to 74% single cells in 8.5μL droplets with Standard Mode. Or for assays requiring lower volumes, dispense with similar efficiency in 2.2μL droplets using Low-Volume Mode (Table 1).
Table 1. Dispense Efficiency2

Pure fluorescent beads were sorted into 3 x 96-well plates with each dispense mode combination. Plates were imaged and single-bead wells were counted. This was completed 3 times for a total of 9 plates/ combination. Efficiency may be higher for cells since bead chemistry makes them likely to stick to plate sidewalls.
PRECISELY amplify low-input DNA and single-cell inputs to reproducibly capture >95% of the genome (Figure 2).
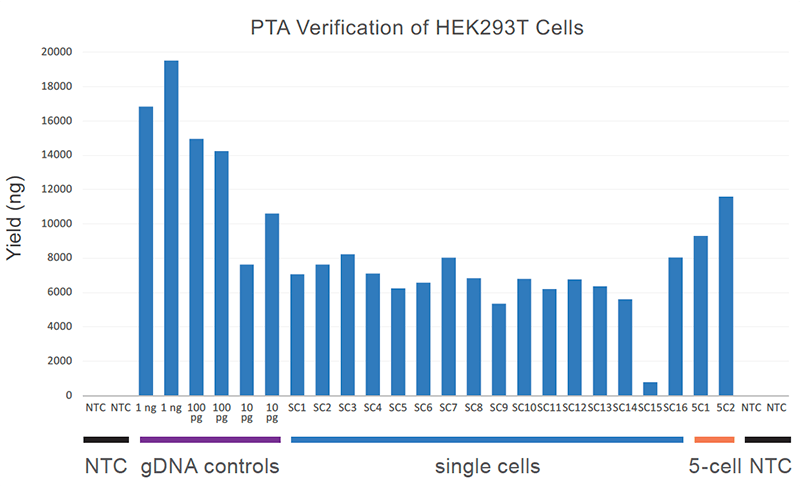
Figure 2. Uniform single-cell yield with PTA: Single-cell amplification reaction yield is shown relative to genomic DNA control and 5-cell input yields. Importantly, no-template control (NTC) reactions are not detectable by Qubit.
DIRECTLY copy the primary template with an isothermal polymerase and proprietary termination chemistry that attenuates the size of amplicons. The smaller amplicons do not efficiently amplify, so randomer primers are redirected to the primary template of interest.
UNIFORMLY amplify with high breadth of coverage, few replication errors, and low allelic bias, to accurately call single nucleotide variants (SNV) at the whole genome sequencing (WGS), whole exome sequencing (WES), and small-panel levels.
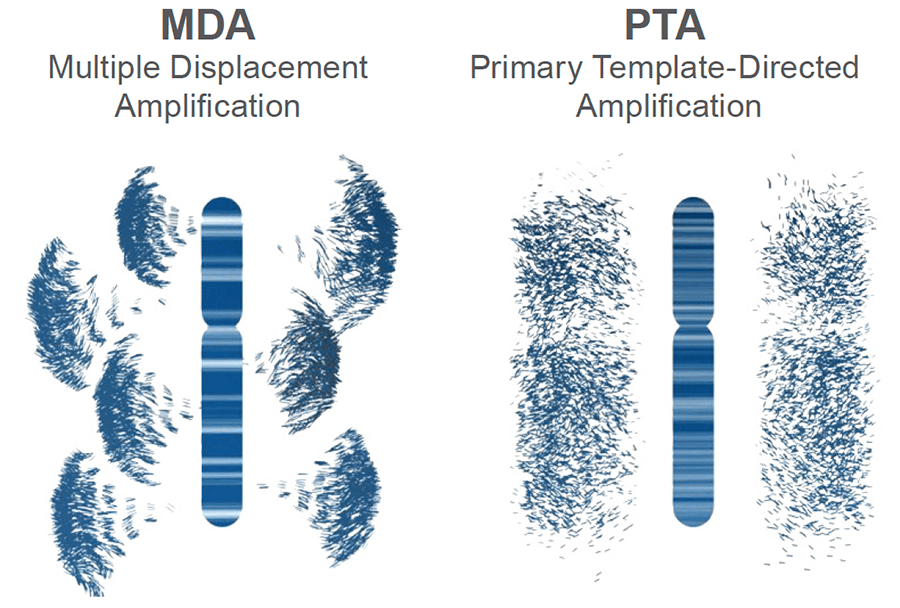
Figure 3. Schematic of genomic amplification output obtained from MDA vs PTA: PTA prevents exponential read pileup and error propagation and yields highly uniform coverage.
ROBUSTLY construct a library using PTA product as input with a choice of library construction protocols. Only 100 ng of unfragmented PTA product is required as input with the ResolveDNA™ Library Preparation Kit from BioSkryb. It uses a ligation-based workflow that does not require fragmentation of the input DNA and utilizes unique dual-index adapters that are compatible with Illumina sequencers.
Unfragmented product can also be used in the KAPA HyperPlus construction protocol, though at a higher 500 ng input. Alternatively, directly tagmented PTA products can be used with Illumina DNA Prep reagents. All workflows yield >400 ng of amplified library, facilitating downstream enrichments if necessary.
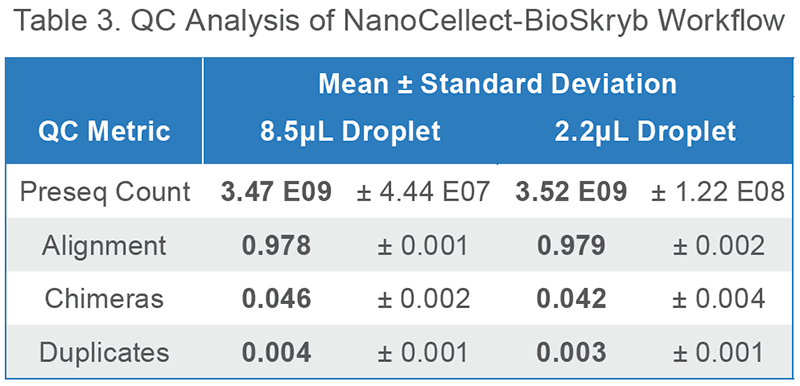
QUICKLY perform low-depth sequencing on any Illumina instrument to obtain QC sequencing metrics prior to performing alignment (Table 3).
SIMPLY analyze high-depth sequencing, align and call variants. and visualize variant call file data with BioSkryb’s BaseJumper Bioinformatics platform (Figure 4). Single nucleotide variation can be explored in the context of a density map–to explore nucleotide differences between single cells at tunable levels of resolution.
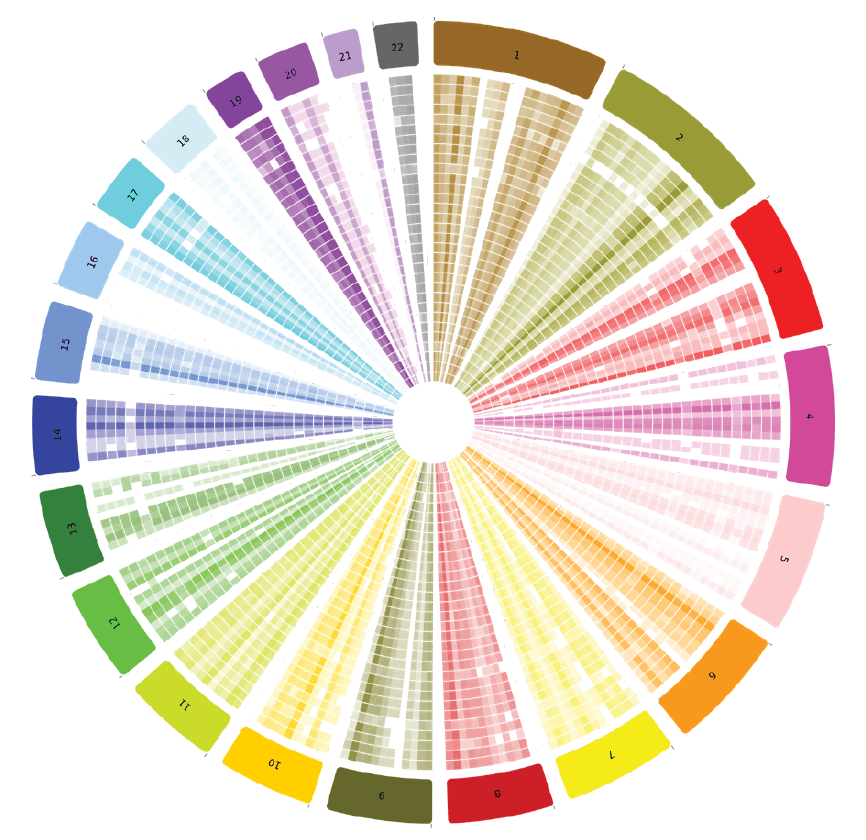
Figure 4. Variant density in BaseJumper: Quickly visually discern genome-wide differences in variant density between single cells (concentric rings) or zoom in for high-resolution regional ascertainment of variant density.
This workflow was verified by sorting live HEK293T cells into 96-well plates with each dispense mode, amplifying with PTA, and low-depth sequencing all wells with >500 ng PTA yield.
All dispense modes yielded even better amplification efficiency than expected from bead testing (Table 2). This is likely due to an improvement in analysis efficiency caused by a difference in particle chemistry – cells may centrifuge to the well bottoms more easily than their sticky bead counterparts. Doublet wells may also contribute to the higher values, since Table 1 bead data only includes singlets, but they do not account for the entire difference; 75-86% of wells contained any number of beads. Therefore amplification yield may often exceed the established bead range.

Viable (SytoxGreen-) HEK293T cells were sorted into 96-well plates with BioSkryb Cell Buffer. 2 total plates were dispensed for each mode. For each plate, PTA was performed on 16 wells and any well with yield > 500 ng was counted.
All single cells yielded a preseq3 library complexity estimate of greater than 3.0E9 bp, which indicates the likelihood of high sensitivity of variant calling upon high depth sequencing. Additional metrics indicate hg38 alignment of > 97% and low chimeric and duplication rate. The data presented here are using KAPA HyperPlus library construction.
Easily amplify single-cell genomes with high breadth, uniform coverage, reduced error propagation, and low allelic dropout by combining NanoCellect’s WOLF and BioSkryb’s ResolveDNA workflow. Accurately call single nucleotide variants (SNV) and copy number variations (CNV) – at the whole-genome, exome, or small panel level – for diverse applications such cancer genomics, prenatal genetic testing (PGT), and neurobiology.
Acknowledgements
Thanks to Adonary Muñoz, Gail Joseph, and Durga Arvapalli for their support on this project.
For more information about single cell multiomics, email [email protected]
References





