Streamlined workflow to integrate microfluidic cell sorting and instrument-free scRNA-seq analysis

Introduction
Single cell RNA-sequencing (scRNA-seq) is a rapidly evolving field with new technologies emerging that increase accessibility, experimental flexibility, and cost efficiency of this powerful method. Reliable and reproducible sample preparation is critically important to the scRNA-seq workflow, and cell sorting methods have been adapted to significantly improve scRNA-seq workflows and experimental design. NanoCellect’s WOLF® and WOLF G2® Cell Sorters can function as a valuable step in the scRNA-seq workflow by isolating only the cells of interest while removing dead cells and debris. This sample enrichment results in significantly higher scRNA-seq resolution of rare cell populations of interest while simultaneously improving sequencing data quality through the removal of dead and damaged cells with degraded RNA. NanoCellect’s unique sorting technology uses disposable and sterile microfluidic cartridges that eliminate the risk of cross contamination between samples. Furthermore, NanoCellect’s cell sorters are more gentle than conventional cell sorters and maintain high post-sort viability and higher RNA integrity. Here, we demonstrate the efficacy of cell sorting on the WOLF G2 upstream of a novel, easily accessible, instrument-free scRNAseq platform developed by Fluent BioSciences.
Fluent BioSciences
Fluent Biosciences offers a breakthrough Pre-templated Instant Partitions (PIPseq™) technology that enables selfassembly of individual cells into partitions without the need of complex instrumentation or expensive consumables. The PIPseq workflow begins by mixing prepared cell suspensions with Fluent’s template particles and generating Pre-templated Instant Partitions (PIPs) by simple vortexing. Cells in PIPs are then lysed on a thermal device and mRNA is captured by barcoded oligonucleotides incorporated on the template particles. cDNA is generated from the captured mRNA via reverse transcription and amplified to create a cDNA library for each individual cell. The single-cell cDNA libraries are then processed with standard library preparation methods and next generation sequencing (NGS) followed by primary data analysis with the Fluent analysis platform (PIPseeker™).
In addition to researchers carrying out their workflow, Fluent BioSciences allows users to ship captured cells (using a Fluent front-end kit) to either the Fluent Service Lab or a validated core lab to perform library preparation, NGS and primary data analysis. This service can be particularly valuable in flow cytometry labs with limited or emerging NGS experience. To demonstrate this workflow, PBMCs and CD4 T cells were processed at NanoCellect’s laboratory and shipped to the Fluent Service Lab for subsequent library preparation, NGS, and analysis.
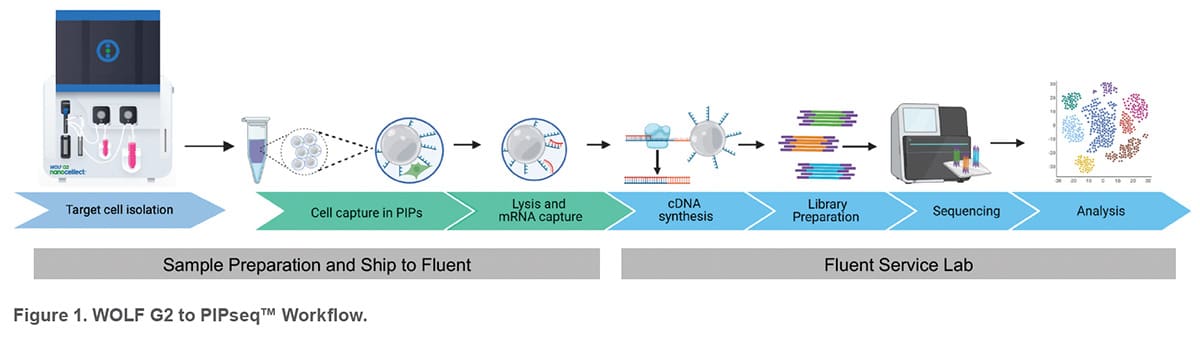
Method
Cryopreserved PBMCs from two donors were thawed and approximately 5,000 unsorted PBMCs from each donor were directly processed into PIPs followed by lysis and mRNA capture. The remaining sample of each donor was stained with CD4-PE (BioLegend #317409), diluted to approximately 800 cells/μL and then stained with the viability dye NucBlue™Live ReadyProbes™ Reagent (Hoechst 33342) (ThermoFisher #R37605). Live CD4+ cells were identified on the WOLF G2 (488/405 nm configuration) and PBS + 0.1% BSA was used as the sample and sheath buffer. 50,000 live CD4+ cells were sorted from each donor and then gently spun down. An estimated 3,000-4,000 of the CD4+ cells were then also processed into PIPs followed by lysis and mRNA capture before being shipped to the Fluent Service Lab.
Once the samples were received by Fluent, the remainder of the scRNA-seq library preparation assay was processed with the PIPseq T2 3’ Single Cell RNA Kit and then sequenced on an Illumina Nextseq 2000 instrument. Analysis was performed by demultiplexing the resulting fastq file and processing through the Fluent proprietary analysis pipeline PIPseeker (v0.52). This software package includes barcode error correction, read mapping, and cell calling, outputting a filtered gene expression matrix. Filtered matrices were processed using the Seurat (v4.0.1) workflow, which contains normalization/transformation/ feature selection by SCTransform, dimensionality reduction by principal component analysis (PCA), graph-based clustering, and visualization using uniform manifold approximation and projection (UMAP). Cell types were identified using SingleR, a tool that compares the RNA signature of each cell to a reference scRNA-seq dataset (in this study, a Seurat-annotated 10x Genomics V1 chemistry dataset).
Results
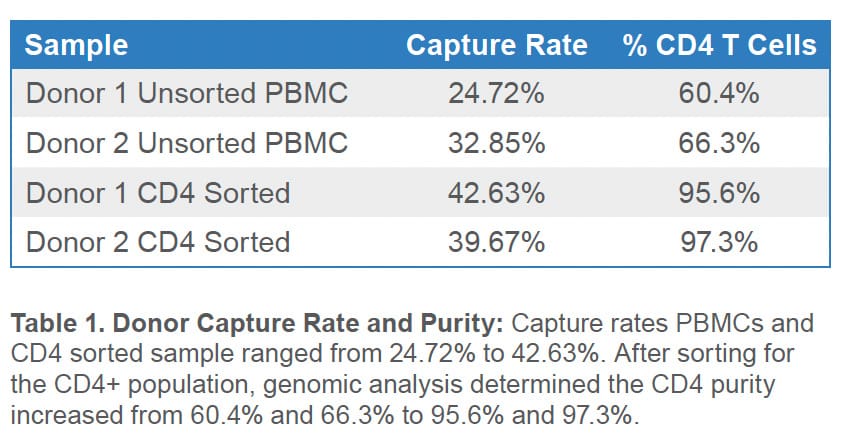
The presence of low viability cells can decrease the rate of high-quality cell capture in scRNAseq experiments because high-quality cells become outnumbered by injured cells with degraded RNA and by cell-free mRNA. This effect is shown by the low cell-capture rate observed for non-sorted cells. Presorting of PBMCs on the WOLF G2 substantially improved the observed cell-capture rates as evidenced by PIPseq analysis. (Table 1).
Population cluster analysis was also performed for unsorted and sorted PBMC samples. Several characteristic immune cell populations were identified in the unsorted PBMC samples (Figure 2). In the CD4 samples, the WOLF G2 was able to enrich CD4 T-cell populations with purities of 95.6% and 97.3% (Table 1 and Figure 2).
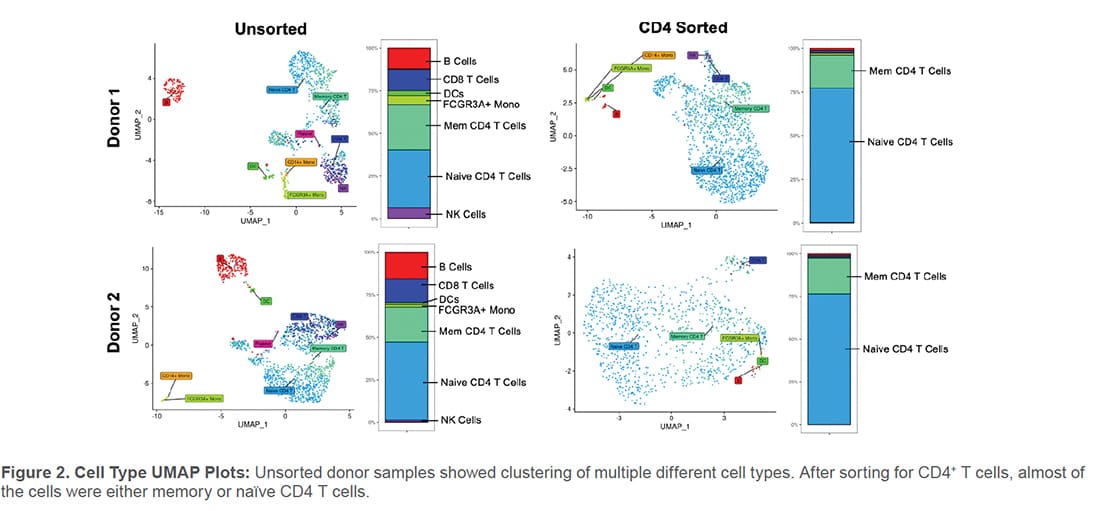
Conclusion
This preliminary demonstration validates the integration of an upstream, gentle cell preparation and target enrichment method on the Nanocellect WOLF G2 Cell Sorter with a highly flexible, instrument-free and cost effective scRNA-seq workflow using the Fluent BioSciences PIPseq T2 3’ Single Cell RNA Kit. This simple, scalable workflow can improve analyzed cell quality and sensitivity by removal of dead cells from samples with compromised viability; and improve the efficiency of sequencing to study rare cell populations of interest. Also, as demonstrated, the ability to conveniently capture cells at the site of cell processing with offsite library preparation and NGS enables powerful scRNA-seq analysis even in labs without significant NGS experience and resources.
For more information, visit nanocellect.com or email [email protected]
Nicole Jagnandan PhD,1 Korina Eribez,1 Pabodha Hettige PhD,2 Aaron May-Zhang PhD2 and Autumn Cholger PhD2
1. NanoCellect Biomedical, Inc., San Diego, CA. 2. Fluent Biosciences, Watertown, MA
WFL – 005